AMPLE can perform Molecular Replacement using a small library of ideal helices. This method requires no modelling and therefore is often very quick.
AMPLE can be found in the CCP4i2 menu under the Molecular replacement tab (shown below)
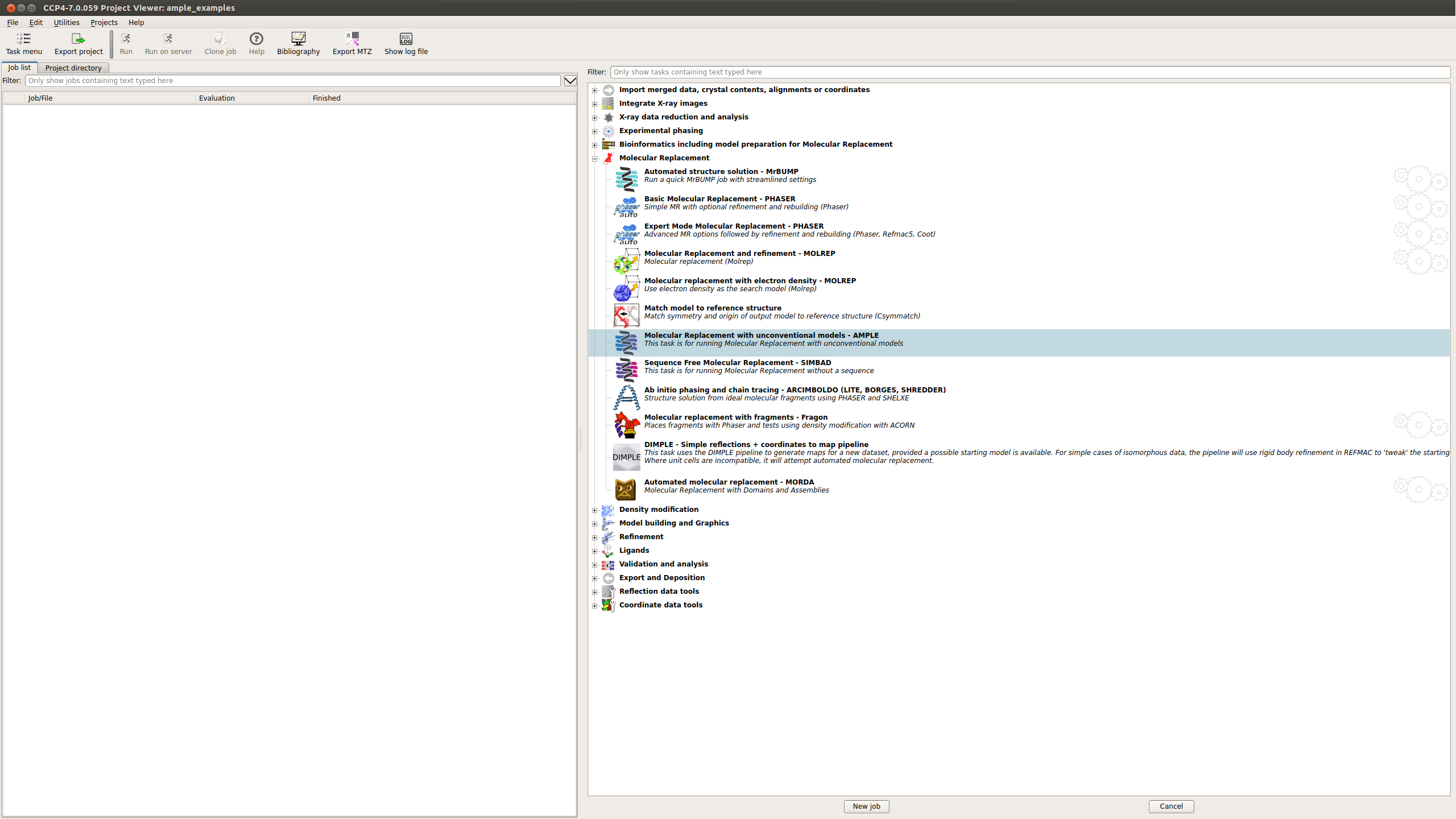
This will take you to the submission page for AMPLE.
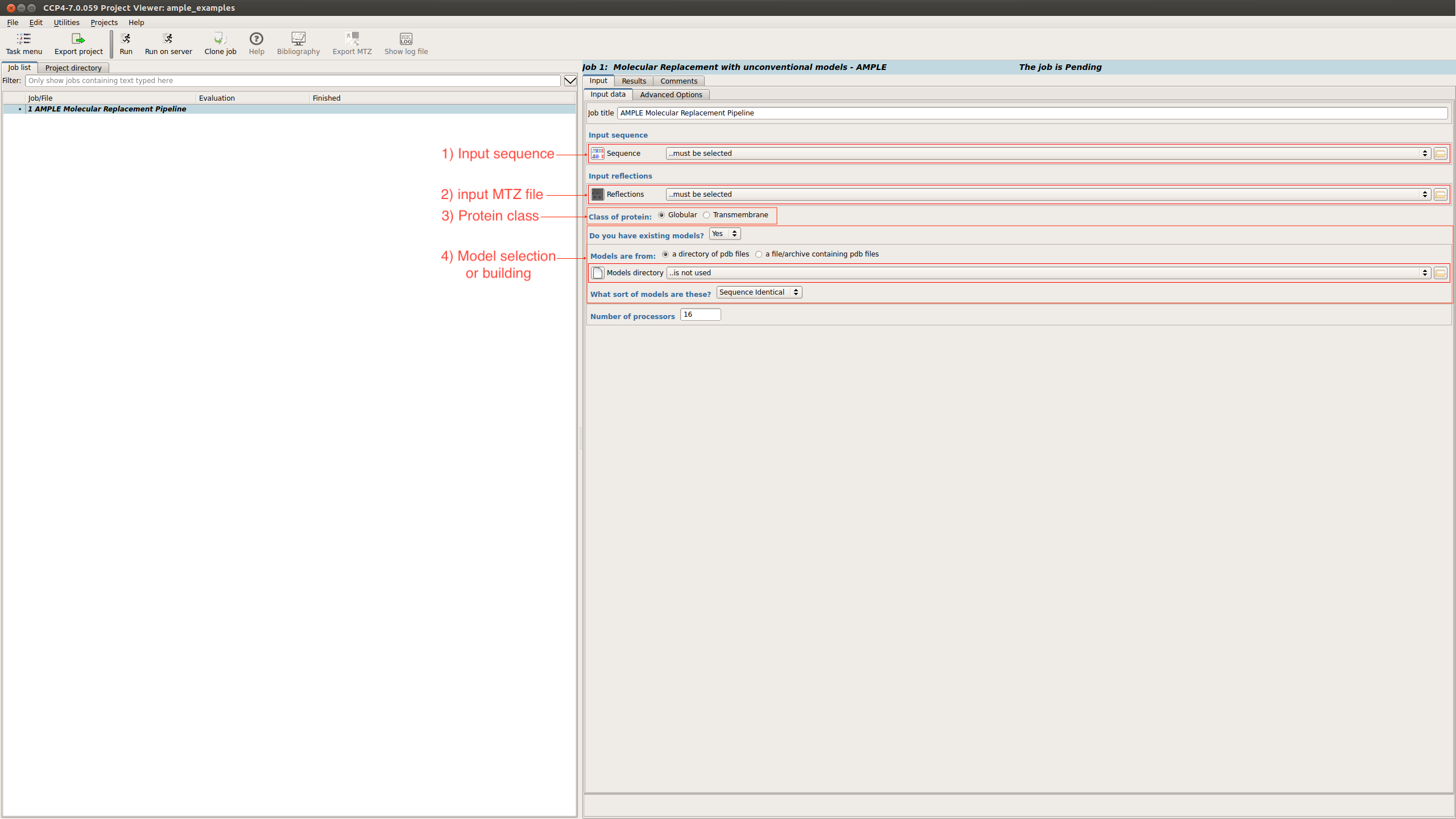
On the submission page there are a number of input options:
There is also an advanced options tab
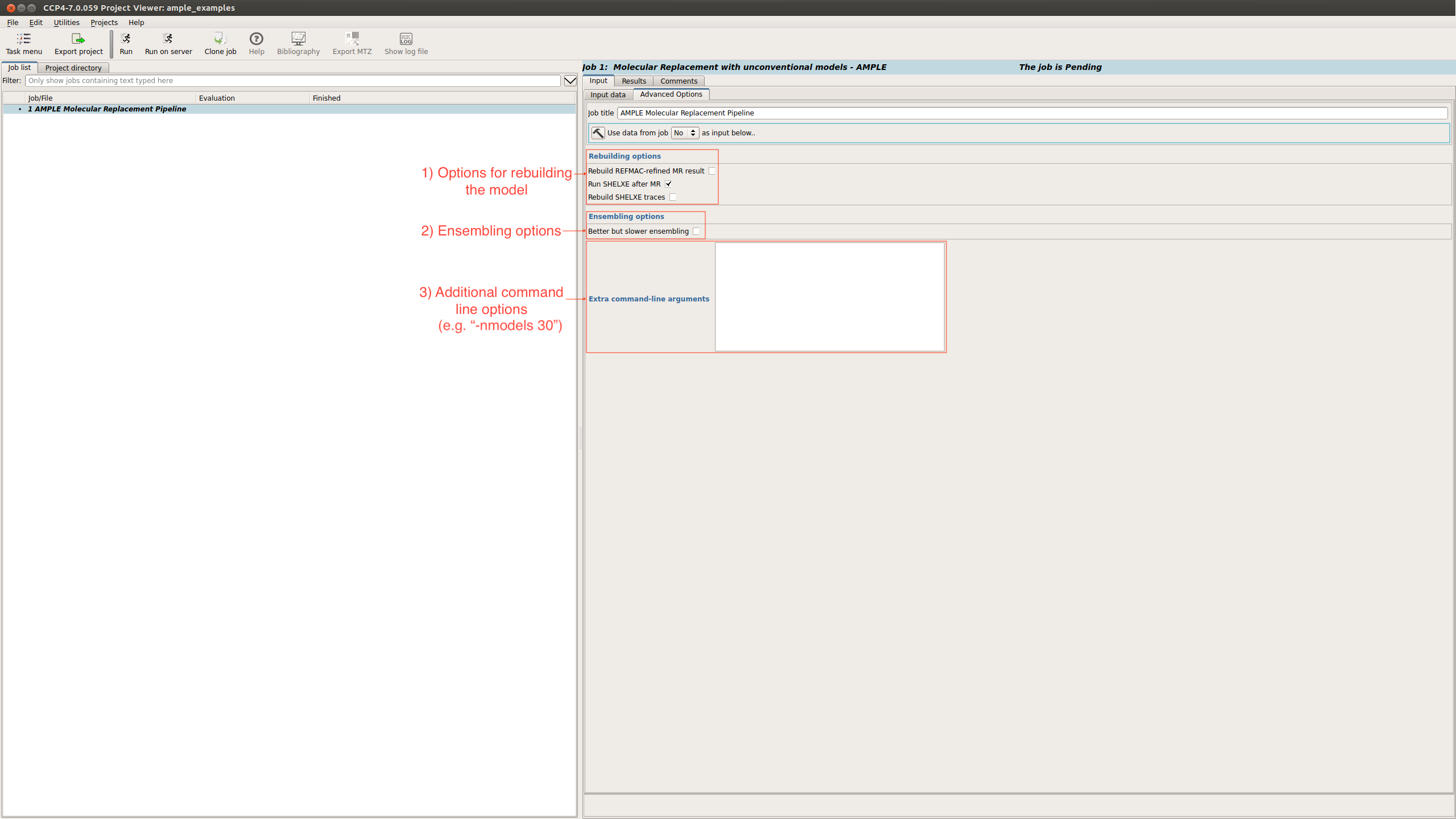
This provides options relating to:
For a full list possible options see AMPLE options.
AMPLE requires a FASTA file and an MTZ file in order to run. There are some other files required, which will be described below.
Note
You can download all the data files here.
The FASTA and MTZ files can be submitted into the fields described above
When using ideal helices the following options need to be input:
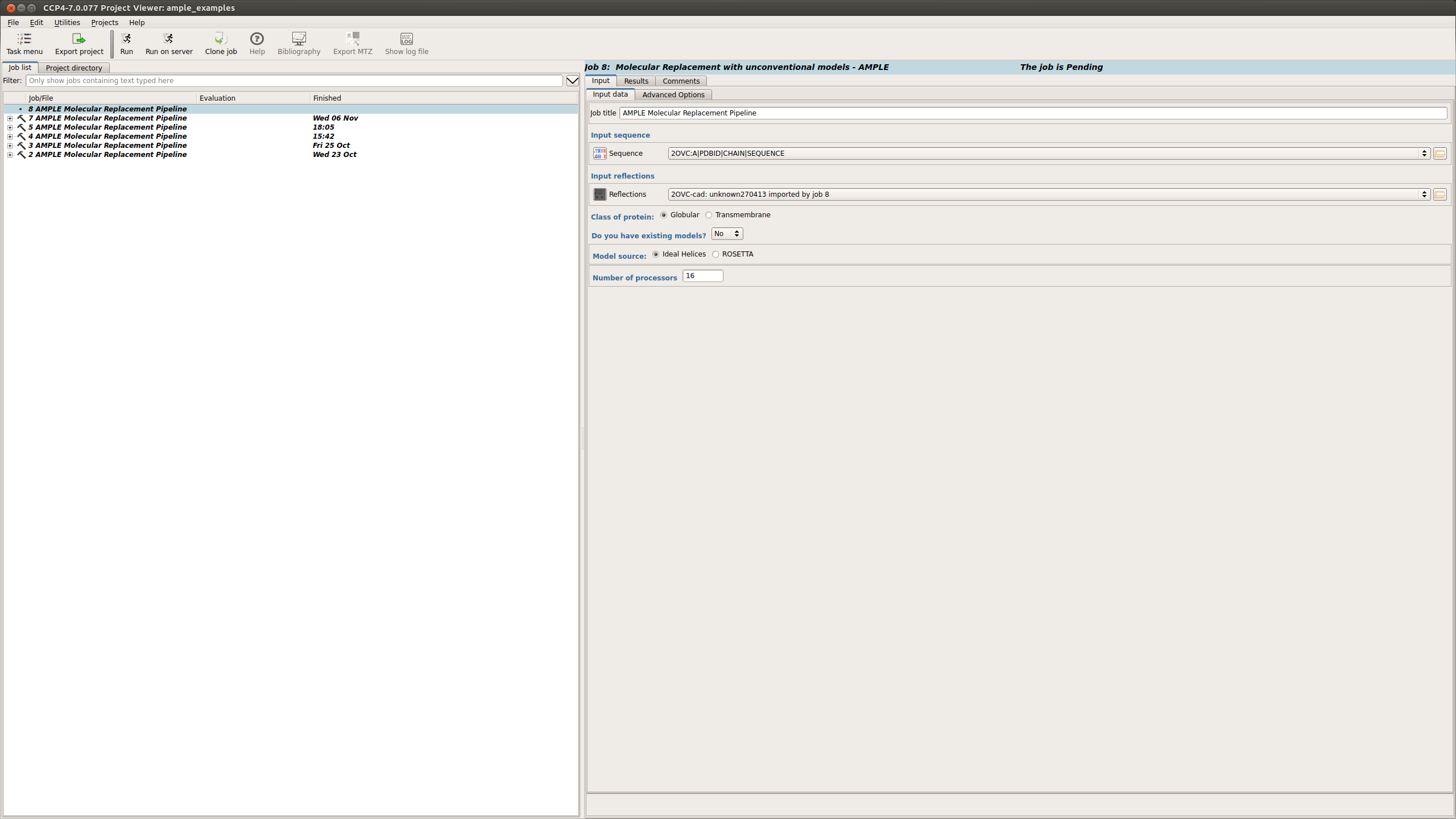
Once these options have been selected the job can be set running.
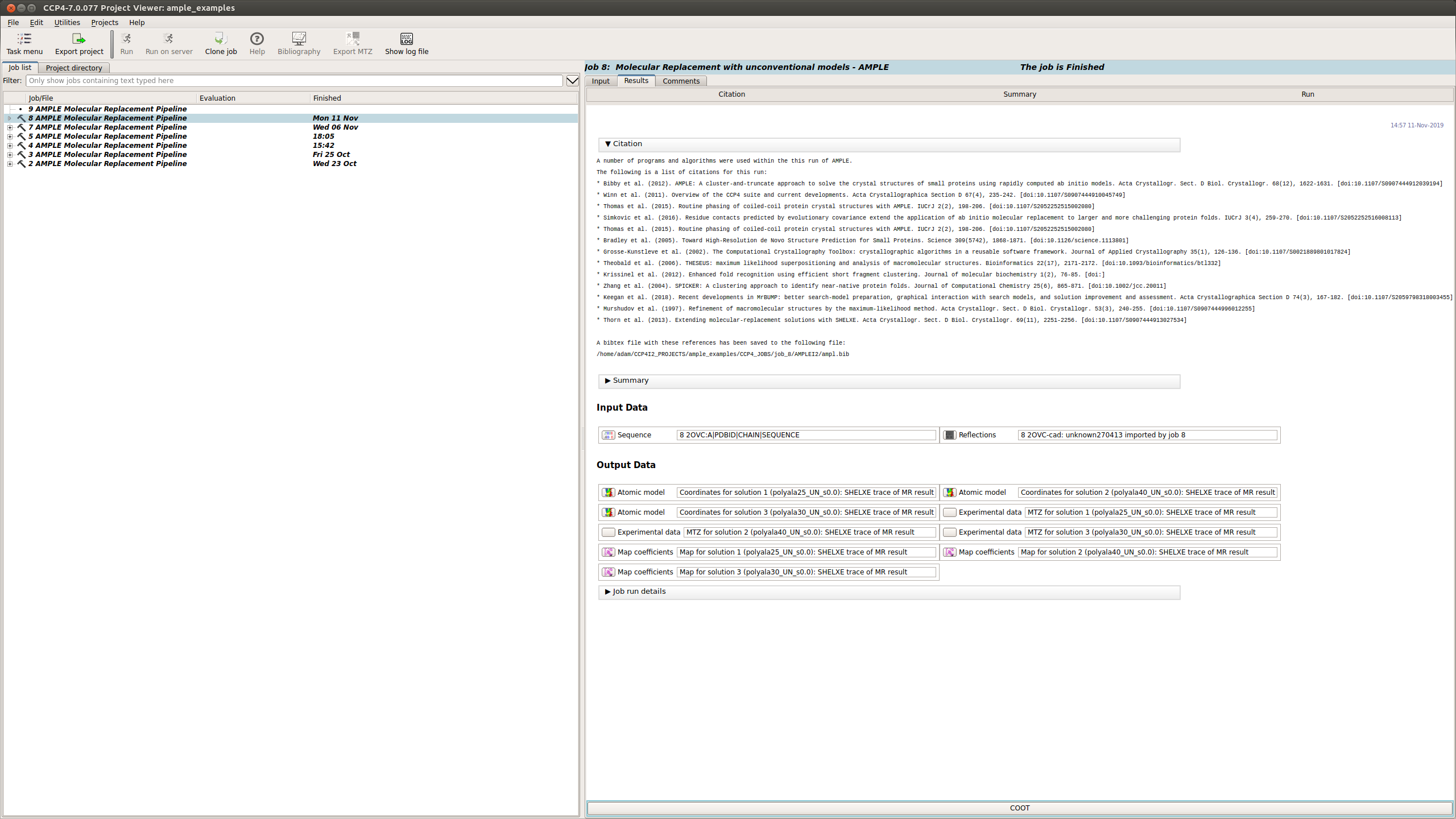
Upon starting a results tab will appear within the CCP4i2 interface summarising the progress of the AMPLE run. This will contain the following sections:
The summary tab contains information about the search from MrBUMP
This section displays a table with the results of running MrBUMP on each of the ensembles, for this example you will have information for the following headings.
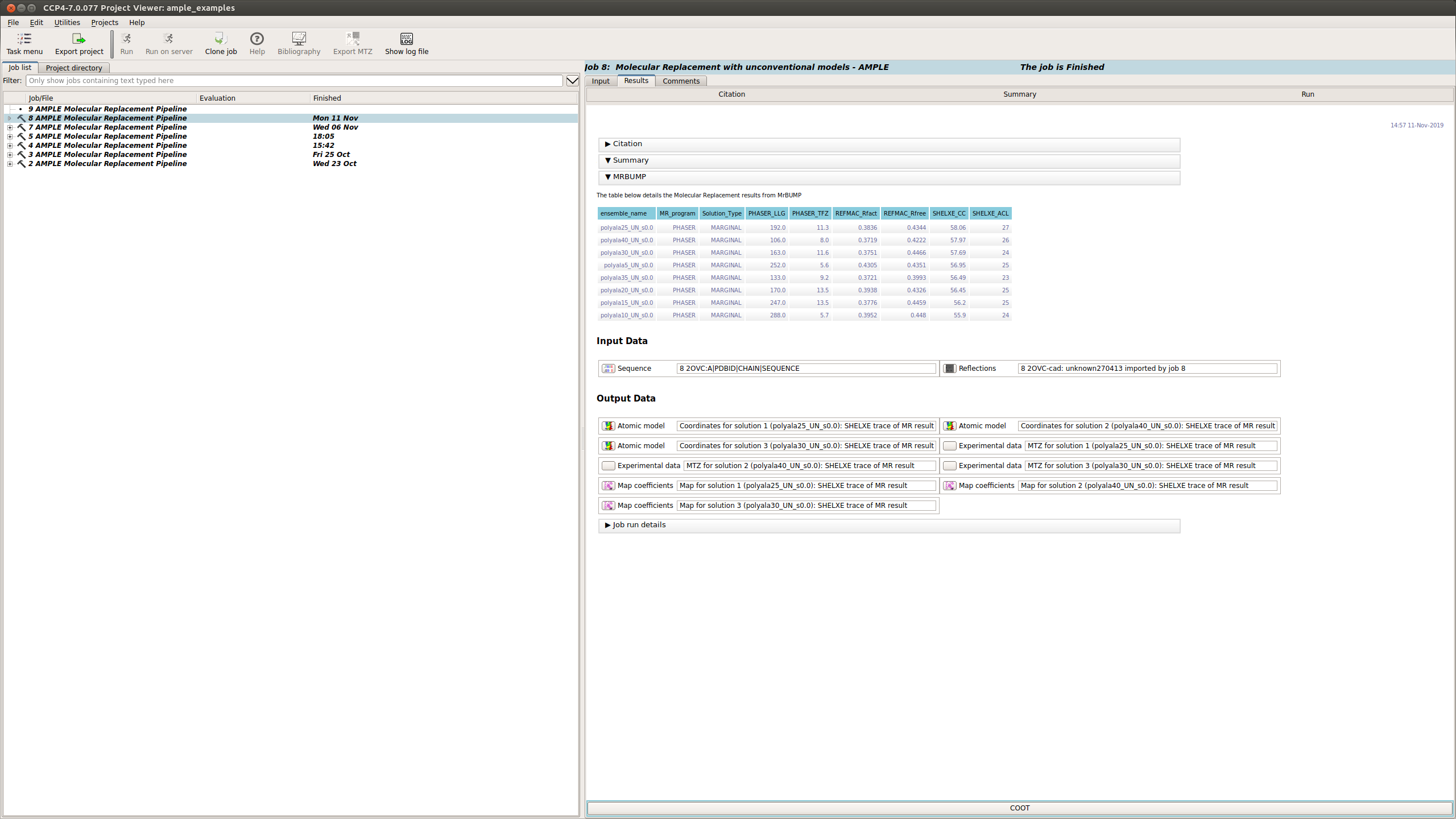
Typically a result with a SHELXE CC score of 25 or higher and a SHELXE ACL of 10 or higher will indicate a correct solution.
The Results tab displays the final results of AMPLE after running MrBUMP on the ensembles.
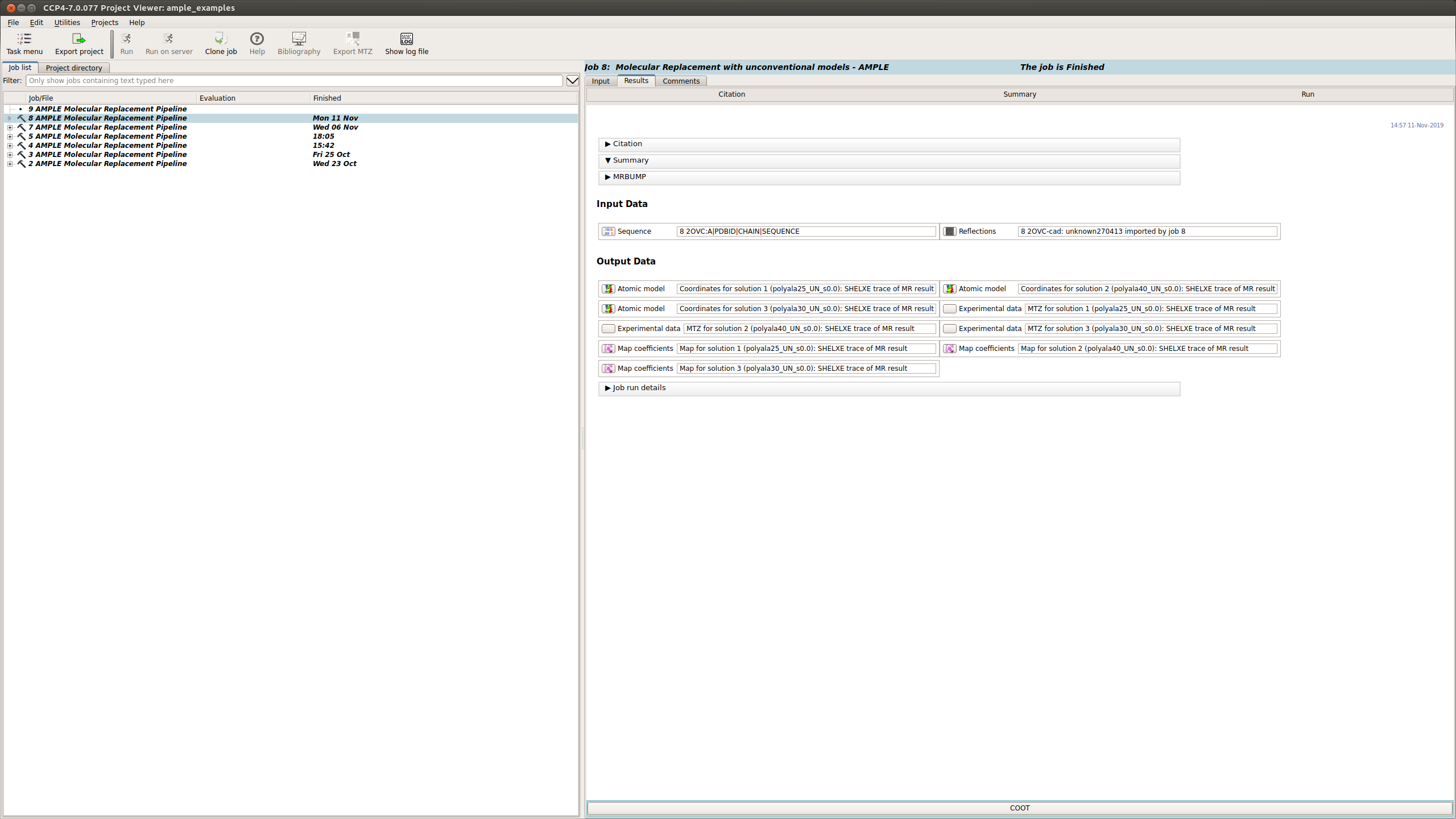
AMPLE output the atomic models, MTZ and map coefficients for the top 3 solutions in the AMPLE run.
Note
The results you obtain may be slightly different to those presented above as you are generating a new slightly different set of ab initio models.
This section lists the programs and algorithms that are using in the AMPLE job and gives a list of references to be cited should AMPLE find a solution.